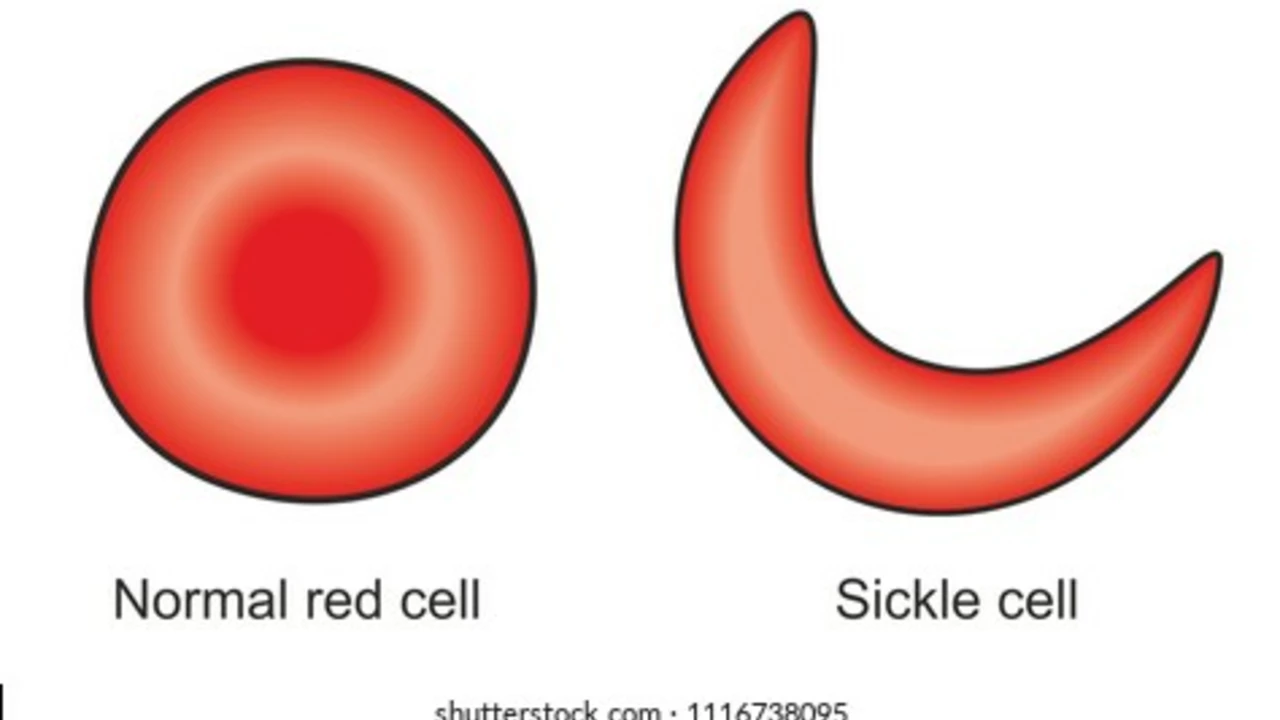
Sickle cell anemia (also called sickle cell disease) is a genetic blood disorder that makes red blood cells stiff and crescent-shaped. Those odd-shaped cells can block blood flow, cause sudden pain, and damage organs over time. If you or someone you care for has sickle cell, knowing the basics helps you act fast when problems start.
The disease comes from inheriting two sickle cell genes — one from each parent. It lowers the red blood cell’s ability to carry oxygen and shortens their life span. Common signs include severe pain episodes (called pain crises), chronic fatigue, jaundice, and more infections than usual. Over time you may see complications like acute chest syndrome, stroke, or organ problems.
Pain can come on fast. First, try simple measures: hydrate, rest, use heat packs on the painful area, and take prescribed pain meds on schedule. Avoid aspirin in young children unless your doctor says so. If pain doesn’t ease within a few hours, or you have fever, trouble breathing, or sudden weakness, get to the ER — these signs can mean a serious complication.
Preventing crises matters. Stay well-hydrated, avoid extreme cold or high altitude, and treat infections quickly. Routine vaccines and yearly flu shots cut dangerous infections that trigger crises.
There are several medicines and procedures that reduce complications. Hydroxyurea is the most used drug — it raises fetal hemoglobin and lowers crisis frequency. Newer options include voxelotor and crizanlizumab, which target different parts of the disease process. Regular blood transfusions help prevent stroke and treat severe anemia. The only widely accepted cure is a bone marrow or stem cell transplant, and gene therapies are showing promise in trials.
Infants diagnosed through newborn screening usually start penicillin and recommended vaccines early to prevent life-threatening infections. Adults need regular checkups to watch for organ damage and to update preventive care.
Living with sickle cell is not just medical care. It’s about planning: know your trigger list, carry a crisis plan and ID card, keep pain meds available, and build a team — hematologist, primary doctor, and trusted local ER. Mental health matters too; chronic pain and hospital stays hit mood and relationships. Seek support groups or counseling when you need it.
Want concise guides or drug information? Look for reliable resources and talk to your care team before changing meds. If you suspect a serious complication — high fever, chest pain, sudden weakness, or severe shortness of breath — go to the emergency room without delay.
Sickle cell anemia is manageable when you know what to watch for and who to call. Small habits—hydration, vaccines, and a clear plan—make a big difference.
© 2026. All rights reserved.